Sau đó Bệnh Andersen là một dạng bệnh dự trữ glycogen đặc biệt nghiêm trọng. Đây là một tình trạng di truyền được đặc trưng bởi sự hình thành glycogen bất thường. Tiên lượng cho bệnh rất xấu.
Bệnh Andersen là gì?

© ag visuell - stock.adobe.com
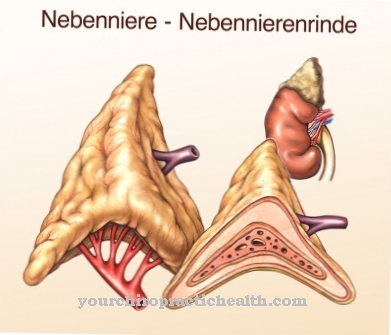
Trong ngữ cảnh của Bệnh Andersen một dạng glycogen bất thường được lưu trữ. Glycogen này có cấu trúc tương tự như amylopectin, một tỷ lệ cao được tìm thấy trong tinh bột thực vật. Thông thường glycogen phân nhánh nhiều. Tuy nhiên, trong bệnh Andersen, chỉ có một polysaccharide phân nhánh yếu.
Bệnh có đặc điểm là gan to ra nhanh chóng, nhanh chóng dẫn đến xơ gan. Polysaccharide bất thường không còn có thể bị phân hủy và tiếp tục tích tụ. Sự thiếu hụt hoặc thậm chí thiếu enzym amylo-1,4-1,6-transglucosidase là nguyên nhân dẫn đến sự hình thành glycogen bị lỗi. Nó cung cấp sự phân nhánh trong phân tử polysaccharide này.
Căn bệnh này rất hiếm gặp, nhưng vẫn xuất hiện ở nhiều dạng hoặc nhiều dạng khác nhau. Ở thể cực kỳ nặng, đứa trẻ thường bị chết lưu. Các dạng nhẹ hơn bắt đầu ở độ tuổi muộn hơn cũng đã được mô tả. Tuy nhiên, trong mọi trường hợp, có một đột biến trong gen (GBE1), nằm trên nhiễm sắc thể số 3.
nguyên nhân
Nguyên nhân của bệnh Andersen là do khiếm khuyết di truyền ở gen GBE1 trên nhiễm sắc thể số 3, gen này có thể di truyền thành tính trạng lặn trên NST thường. Gen này chịu trách nhiệm tổng hợp enzyme amylo-1,4-1,6-transglucosidase. Nếu thiếu enzym này hoặc nếu nó chỉ có chức năng hạn chế, glycogen bình thường không thể được tổng hợp nữa. Enzyme chịu trách nhiệm cho sự phân nhánh của phân tử polysaccharose.
Nếu sự phân nhánh này không diễn ra hoặc nếu nó chỉ được thực hiện không hoàn toàn, một glycogen sẽ được tạo ra mà không còn có thể bị phân hủy để cung cấp năng lượng nhanh chóng. Ngược lại, nó tích tụ rất nhanh trong gan, lá lách và các hạch bạch huyết. Sau mỗi bữa ăn, một phần glucose chưa được sử dụng sẽ được vận chuyển đến gan để dự trữ dưới dạng chất dự trữ là glycogen.
Tuy nhiên, vật liệu dự trữ này không thể được sử dụng ở dạng hiện tại. Sự tích tụ liên tục của glycogen bất thường khiến gan và lá lách ngày càng to ra và chắc chắn dẫn đến sự hủy hoại của cả hai cơ quan.
Các triệu chứng, bệnh tật & dấu hiệu
Bệnh Andersen biểu hiện qua một sự biến đổi bất thường. Đó là về việc lưu trữ liên tục một glycogen bất thường, không thể phân hủy được nữa. Nhưng mức độ nghiêm trọng của bệnh có thể khác nhau. Tuy nhiên, tiên lượng về bệnh Andersen nói chung là rất xấu. Triệu chứng nổi bật nhất là gan to liên tục, từ đó bệnh xơ gan nhanh chóng phát triển.
Dạng nặng nhất thể hiện qua việc trẻ bị thiếu hoặc giảm các cử động trước khi sinh. Thai nhi có dấu hiệu cứng khớp và thiểu sản phổi. Thông thường trong những trường hợp này đứa trẻ sinh ra đã chết. Trong những trường hợp cổ điển, đứa trẻ vẫn phát triển bình thường khi sinh ra. Tuy nhiên, trong vài tháng đầu đời, gan to (gan to) và giảm trương lực cơ (thiếu căng cơ) phát triển.
Nhìn chung, sự phát triển của trẻ bị chậm lại. Bệnh tiến triển nhanh chóng. Gan phát triển thành xơ gan. Ngoài ra còn có tăng áp lực cửa và lá lách to ra. Do xơ gan, các tĩnh mạch phát triển trong thực quản kèm theo xuất huyết và cổ trướng tương ứng. Tử vong thường xảy ra trong thời thơ ấu. Trong một số trường hợp hiếm hơn, bệnh khởi phát muộn hơn và xuất hiện các triệu chứng yếu cơ và suy tim. Các triệu chứng thần kinh cũng xảy ra ở đây.
Chẩn đoán & diễn biến bệnh
Chẩn đoán có thể được thực hiện trên cơ sở hình ảnh lâm sàng và kèm theo các xét nghiệm cận lâm sàng, sinh thiết gan và xét nghiệm di truyền phân tử. Trong các xét nghiệm mô học, sự tích tụ nội bào của các cấu trúc giống như amylopectin có thể bảo quản được là đáng chú ý. Enzyme chịu trách nhiệm được kiểm tra trong tế bào gan, nguyên bào sợi và bạch cầu. Sự thiếu hụt amylo-1,4-1,6-transglucosidase đã được chứng minh xác nhận chẩn đoán.
Các biến chứng
Theo quy luật, tuổi thọ của đứa trẻ bị giảm đáng kể bởi bệnh Andersen hoặc đứa trẻ sinh ra đã chết. Điều này có thể dẫn đến những phàn nàn hoặc trầm cảm về tâm lý, đặc biệt là với người thân hoặc cha mẹ. Trong hầu hết các trường hợp, họ phụ thuộc vào điều trị tâm lý.
Trẻ em bị ảnh hưởng bị xơ gan, cuối cùng dẫn đến tử vong. Hơn nữa, các khớp cũng bị cứng lại và không thể cử động được nữa do phàn nàn này. Sự phát triển tinh thần của đứa trẻ cũng bị suy giảm nghiêm trọng bởi căn bệnh Andersen, do đó những người bị ảnh hưởng thường phải phụ thuộc vào sự giúp đỡ của người khác. Không hiếm trường hợp suy tim hoặc yếu cơ xảy ra.
Người bệnh cũng có thể chết vì chết tim. Thật không may, bệnh của Andersen không thể chữa khỏi. Việc cấy ghép gan cũng chỉ có thể làm giảm các triệu chứng trong một thời gian ngắn, vì gan mới cũng sẽ bị tổn thương. Điều này cuối cùng dẫn đến cái chết của đứa trẻ. Tuy nhiên, cho đến lúc đó, các phàn nàn và triệu chứng có thể được hạn chế với sự trợ giúp của các biện pháp y tế.
Khi nào bạn nên đi khám?
Bệnh Andersen là một bệnh di truyền, trong trường hợp nghiêm trọng, có thể dẫn đến cái chết của thai nhi trong bụng mẹ. Vì vậy, thai phụ nên đi khám và điều trị ngay khi nhận thấy những biểu hiện bất thường hoặc bất thường trong thai kỳ. Nếu bà mẹ tương lai có cảm giác mơ hồ rằng có thể có điều gì đó không ổn với thai nhi, cô ấy nên hỏi ý kiến bác sĩ. Nếu trẻ sơ sinh sống sót trong vài ngày và vài tuần đầu tiên sau khi sinh, thì cần đến bác sĩ ngay khi các điểm đặc biệt trở nên rõ ràng trong quá trình phát triển tiếp theo. Nếu bạn bị yếu cơ hoặc rối loạn vận động, nên hỏi ý kiến bác sĩ.
Rối loạn tăng trưởng là dấu hiệu của một căn bệnh hiện có và phải được làm rõ. Phải khám và điều trị những bất thường về tim, dị dạng cơ thể và bất thường về hành vi của trẻ. Trong nhiều trường hợp, bệnh dẫn đến mở rộng các cơ quan. Gan hoặc lá lách nói riêng bị ảnh hưởng trong những trường hợp này.
Do đó, cần đến bác sĩ ngay khi xuất hiện hình dạng bất thường của phần trên cơ thể so với trẻ sơ sinh hoặc trẻ cùng tuổi. Da đổi màu hoặc các biểu hiện bất thường khác trên da là những dấu hiệu của sự suy giảm sức khỏe. Mặt hoặc mắt hơi vàng nên được bác sĩ đánh giá.
Trị liệu & Điều trị
Vì bệnh có tính chất di truyền nên không thể điều trị theo nguyên nhân. Liệu pháp chỉ mang tính chất triệu chứng. Là một phần của điều trị, các bác sĩ chủ yếu tập trung vào các biến chứng phát sinh. Điều này làm giảm áp lực trong mạch tĩnh mạch cửa. Ngoài ra còn có sự thay thế của albumin và các yếu tố đông máu.
Nếu bị suy gan, ghép gan có thể kéo dài sự sống. Tuy nhiên, căn bệnh này không thể chữa khỏi ngay cả khi được ghép gan. Khiếm khuyết di truyền hiện diện và cũng sẽ dẫn đến lắng đọng glycogen bất thường trong gan mới. Việc lưu trữ polysaccharide bị lỗi vẫn tiếp tục trong các cơ quan khác của cái gọi là hệ thống tế bào lưới của lá lách và các hạch bạch huyết, do đó các biến chứng nghiêm trọng vẫn có thể xảy ra ngay cả sau khi ghép gan thành công.
Hệ thống tế bào lưới là một phần của hệ thống miễn dịch và bao gồm các tế bào của mô liên kết dạng lưới. Những tế bào này lưu trữ các hạt và chất để phá vỡ chúng và sau đó mang chúng ra khỏi cơ thể. Tuy nhiên, việc phân hủy các phân tử polysaccharose bị lỗi cũng không thể xảy ra ở đây nữa.
Triển vọng & dự báo
Bệnh Andersen có tiên lượng tương đối xấu. Căn bệnh chuyển hóa cho đến nay vẫn chưa thể chữa khỏi và khiến gan bị tổn thương nghiêm trọng. Trong một số trường hợp, các triệu chứng về cơ và các bệnh đồng thời xảy ra, nếu không được điều trị sẽ tiến triển nặng dần. Tuổi thọ bị giới hạn nghiêm trọng bởi điều kiện. Trẻ em bị bệnh đạt trung bình từ hai đến năm tuổi. Ghép gan sớm giúp cải thiện tiên lượng. Tiên lượng xấu, đặc biệt đối với các dạng cổ điển của bệnh, đặc biệt nếu không được ghép gan trong vài tháng đầu đời.
Theo quy luật, tiên lượng lâu dài dựa trên mức độ, mức độ nghiêm trọng và sự tiến triển của bệnh. Bệnh Andersen là một trong những bệnh glycogenose nghiêm trọng nhất. Chất lượng cuộc sống thường giảm đi rất nhiều do các vấn đề về gan và các triệu chứng khác. Thuốc giảm đau và liệu pháp toàn diện cải thiện sức khỏe của trẻ, nhưng chúng cũng đi kèm với rủi ro. Các bác sĩ chuyên khoa gan chịu trách nhiệm cung cấp tiên lượng.
Tuổi thọ bị giới hạn nghiêm trọng bởi điều kiện. Bất kỳ bệnh đồng thời nào có thể xảy ra với các bệnh chưa được phát hiện cũng được đưa vào tiên lượng. Do đó, bệnh Andersen có tiên lượng xấu về tổng thể. Các phương pháp điều trị mới có thể mang lại sự cải thiện trong tương lai.
Phòng ngừa
Phòng ngừa bệnh Andersen chỉ có thể đề cập đến thực tế là con cái không bị di truyền căn bệnh này. Vì bệnh Andersen di truyền theo kiểu lặn trên NST thường, nên di truyền có thể bỏ qua một số thế hệ.Nếu trong gia đình và người thân đã có người mắc bệnh Andersen, thì nên tiến hành xét nghiệm gen người.
Nếu gen được tìm thấy ở cả bố và mẹ, thì nên tư vấn di truyền. Trong trường hợp này, con cái có 25% nguy cơ mắc bệnh Andersen.
Chăm sóc sau
Vì bệnh Andersen không thể chữa khỏi nên việc điều trị các triệu chứng và ngăn chặn các biến chứng có thể xảy ra là trọng tâm chính trong suốt thời gian điều trị. Chăm sóc theo dõi là cần thiết sau các can thiệp được thực hiện như một phần của liệu pháp. Nếu một ca ghép gan xảy ra, việc chăm sóc theo dõi chuyên nghiệp là rất quan trọng đối với nó.
Sau thủ thuật, điều này đảm bảo rằng gan mới không bị cơ thể từ chối. Thuốc đặc biệt ngăn chặn phản ứng miễn dịch của cơ thể. Tuy nhiên, kết quả là sức đề kháng của cơ thể đối với các tác nhân gây bệnh bị suy yếu, điều này phải được tính đến khi điều trị thêm. Trong thời gian này, bệnh nhân phải xét nghiệm máu thường xuyên. Cẩn thận để đảm bảo rằng không có phản ứng đào thải hoặc các biến chứng nghiêm trọng khác như rối loạn chức năng thận, có thể xảy ra như tác dụng phụ.
Trong khi các triệu chứng cốt lõi của bệnh Andersen có thể được cải thiện trực tiếp sau khi ghép gan, sự lắng đọng glycogen bị lỗi vẫn tiếp tục, do đó, các biến chứng và các triệu chứng tiến triển phải được dự kiến ngay cả sau khi cấy ghép. Bác sĩ chuyên khoa gan chịu trách nhiệm có thể cung cấp thêm thông tin chi tiết về tiên lượng và quá trình điều trị.
Bạn có thể tự làm điều đó
Các biện pháp tự trợ giúp mà một bệnh nhân mắc bệnh Andersen có thể thực hiện được giới hạn ở mức không tồn tại. Vì căn bệnh này có nguyên nhân di truyền và không thể kiểm soát được mặc dù đã được điều trị triệu chứng nên khả năng của người bị ảnh hưởng nhanh chóng trở nên cạn kiệt. Tốt nhất anh ta nên thực hiện mọi lời khuyên về chế độ ăn uống và lối sống do bác sĩ điều trị đưa ra một cách nghiêm túc và thực hiện nó.
Hơn nữa, sau khi ghép gan, người bị ảnh hưởng nên cân nhắc hành vi nhẹ nhàng. Nên tránh rượu, thức ăn béo và gắng sức. Điều này giúp cơ thể thực sự chấp nhận cơ quan mới dễ dàng hơn. Tuy nhiên, một ca cấy ghép thành công, bao gồm cả việc chăm sóc theo dõi thành công, không thể tự ngăn chặn bệnh glycogenosis loại 4.
Vì đây là một bệnh di truyền lặn trên NST thường (có thể bỏ qua vài thế hệ), nên việc lập hồ sơ di truyền khi lập kế hoạch gia đình là rất hợp lý. Trong khi những người bị ảnh hưởng bởi bệnh Andersen đã biết về gen của họ, một phân tích về vấn đề này đặc biệt đáng giá đối với các thành viên trong gia đình. Bằng cách này, có thể ngăn chặn việc truyền gen kích hoạt thông qua kế hoạch hóa gia đình phù hợp. Tuy nhiên, ít nhất có thể có được một sự chắc chắn về nguy cơ mắc bệnh ở con cái của chính mình.

.jpg)
.jpg)

.jpg)
.jpg)




.jpg)
.jpg)